优养在线
优养在线高端医疗器械创新遇审批中梗阻 企业谁创新谁倒霉
为增进我国医疗技术创新,最近几年来国家加快实行药品、医疗器械审批制度改革。但记者近期在多家高端医疗器械生产企业采访时发现,产品注册耗时长、审批标准不明晰、部份环节冗繁、物价审核滞缓等问题仍然存在。注册、物价审批“中阻塞”致使部份企业的医疗器械新产品、新技术进入临床利用周期太长影响国产品牌竞争力,1些企业被迫放弃国内市场去拓展海外市场,1些企业耗费巨资研发的技术难以转化堕入破产地步。
高端医疗器械遇审批“中阻塞”
药品、医疗器械审批耗时长、环节多是困扰医疗行业的老大难问题,多家企业向记者表示,高端医疗器械新产品两3年才能完成注册仍然普遍。
1家企业负责人说:“近几年相干部门对医疗器械管理日益严格,这是好事,但是审批时间也在增加。我公司的1款产品2013年申报注册,2014年上半年就审批完成。但这款产品的改进型从2014年下半年申报,到现在还没有拿到批文。”
另外一家企业的首席技术官说,高端医疗器械多为3类医疗器械,注册由国家相干部门审批,全国送审的产品数量众多且触及的学科门类复杂,而审评人员10分有限,对1些新技术其实不了解,从而构成审批“堰塞湖”。“2015年我公司的1款产品到期续证,还不是新产品注册,就花了1年多时间。这款产品1年的销售额就是1亿元。”
针对企业反应的问题,2014年相干部门制定了创新医疗器械特别审批程序,为新技术的推行利用开设审批“绿色通道”。但是即便取得特别审批,产品注册也并不是1帆风顺。
我国心衰患者约450万人,但年心脏移植手术仅300例左右,为弥补我国人工心脏空白,西部1家企业引进国外技术研发人工心脏产品。“目前人工心脏在日本、美国售价超过100万元1套,国产化后可下降到40多万元,”该企业负责人对这1项目充满信心,并于2015年底取得特别审批资历。原以为产品会如期上市,但由于中途国内新标准引入、产品补充检测仍在等待,1年多过去了临床实验还未启动,产品什么时候上市更是难以预知。
除审评气力不足外,审批标准不明晰、部份环节冗繁也是注册审批耗时长的重要缘由。1家企业负责人说,医疗器械新技术不可能完善,而审批是不是合格又没有明确标准,审评人员有时对产品风险过于苛求,常常提出问题又拿不出法律政策根据,“我们有1款产品在审批时被要求补充资料10屡次,至今还没有完成审批。而这款产品在美国FDA只补充了1次资料,8个月就完成了注册审批,拿到欧盟的批文只用了3个月。”
依照相干规定,医疗器械临床实验应当通过实验机构伦理委员会的审议。1家企业负责人说,医院伦理委员会1般两至6个月开1次会,每次审议的实验项目有限,为提高时效,1些企业不能不花钱、找关系去公关。
即便企业拿到了产品注册证,进入医院临床利用还面临物价审核。我国非营利性医疗机构的医疗服务实行政府指点价,医疗器械新产品的收费须由公立医院向当地物价、卫生、社保部门申请。“因物价和医保政策差异,各地物价审核履行力度不1。每一个省、地级市都要去做工作,多数地方对新产品的物价审核严重滞后,简直要把企业跑死累死”,受访企业负责人说。
企业“谁创新谁倒霉”
我国高端医疗器械起步晚、技术新、投入大、风险高,又面临国外品牌的竞争压力,及时上市对科技成果转化、国产品牌抢占市场具有重要意义,而这些产品注册和物价审批中存在的“中阻塞”严重制约了我国高端医疗器械的创新和发展,受访企业负责人无奈地说,“谁创新谁倒霉”。
上述研发人工心脏的企业负责人介绍,国内人工心脏尚属空白,缺少技术标准和检测装备,检测和临床实验耗时长、费用高达数千万元,作为中外合资公司,该公司引入的国外技术取得美国临床实验豁免并已在欧美上市,公司建议参照国际惯例对引入型成熟技术减免他国已做过的测试项目。国内相干专家认可公司建议,但相干审批部门对此1直未给出明确答复,审批进展缓慢。该企业自2014年成立至今已投入8000多万元,目前面临外资准备中断合作、公司濒临破产倒闭的窘境。
因审批繁琐漫长,1些企业不能不放弃国内市场去拓展海外市场。1家企业负责人介绍,该企业目前正在销售的20多个产品中,只有20%拿到了国内注册和物价批文,海外市场占到了公司60%的份额。即便在欧盟和美国监管部门已注册,想在国内销售仍需进行临床实验,而多数国外产品则不用。同时,2015年12月国家发改委专门发文要求各地加快受理新增医疗服务价格项目,但1年多过去了,多数地方依然未动,新研发的产品只能置之不理,极大地打击了科研人员的积极性。“我国医疗市场10分巨大,但是审批‘拦路虎’让我们进入国内市场困难重重,反倒让给了国外品牌。”
加快审批制度改革的落实
针对注册、物价审批“中阻塞”,受访企业负责人建议应加快医疗器械审批制度改革的落实,买通国产高端医疗器械上市“最后1千米”。
首先,可鉴戒美国、欧盟管理经验,试点3类医疗器械检测、评审、认证分立,疏通注册积存“堰塞湖”。受访企业负责人介绍,欧美的申请注册的高端医疗器械数量很多,但未出现积存问题是由于将产品检测交由专业的市场化检测机构,乃至企业可自检,并制定有极其严格的监管措施,1旦检测机构和企业造假将面临严厉处罚。他们建议,可授权更多第3方检测机构检测3类医疗器械,技术审批可试点下放到部份省市,最后由国家相干部门把关认证。
再次,明确审批人员权责,优化审批环节。受访企业负责人建议,应进1步明确注册审批细则、标准、透明度和审批人员的权限和职责,审批人员提出疑问应有明确政策根据,限制补充资料次数,只要审批人员依照相应程序审批可免于责任,避免“谁审批谁负责”影响审批人员积极性;同时,医院伦理委员会应重点保障临床实验者权益,对3类医疗器械临床实验的有效性应由省级管理部门组织专家进行评估。
第3,统1受理新增医疗服务价格审核。受访企业负责人建议,对3类医疗器械的医疗服务价格审核和是不是进入医保目录,应归口到指定部门,该部门审定后,各省市可根据当地情况在1定范围内调剂后统1履行。从长远来看,还应改革医疗服务价格政府定价,逐渐实现市场化。
-
LifeSpace荣获第62届澳大利亚出口奖"电子商务"类大奖
广州2024年11月26日 /美通社/ -- 近日,第62届澳大利亚出口奖颁奖典礼在澳大利亚堪培拉的议会大厦举办,全球领先专...[详细]
-
波士顿科学宣布达成Intera Oncology® Inc.医疗器械公司收购协议
——该收购为肿瘤介入产品组合新增了治疗肝转移瘤的补充疗法 马萨诸塞州马尔伯勒2024年11月26日 /美通社/ -- 近日...[详细]
-
诺和诺德亮相第二届链博会:以"链"会友,共筑健康未来
北京2024年11月26日 /美通社/ -- 今日,第二届中国国际供应链促进博览会(简称"链博会")在北京盛大开幕。...[详细]
-
富士胶片与上海理工大学合作医疗器械实践教育基地正式揭牌
上海2024年11月26日 /美通社/ -- 11月20日,上海理工大学医疗器械校外实践基地在富士胶片智慧医疗技术培训中心揭牌...[详细]
-
AsiaPay为亚太区女性领导企业提供4万澳元奖金
连续第二年支持女性创业者推动积极变革 澳大利亚墨尔本2024年11月26日 /美通社/ -- 为女性创业者赋能的全球平台...[详细]
-
赛诺菲"奇迹紫赛道"亮相第二届链博会
追寻免疫健康链路,点亮生命赛道 北京2024年11月26日 /美通社/ -- 全球领先的创新医药健康企业赛诺菲首次参展第二...[详细]




-
-
卫材抗癌药物"TASFYGO®片剂35mg"在日本上市
用于治疗携带FGFR2基因融合或重排的胆道癌 东
-
对您来说,干净的洗手间有多重要? 最新全球调查揭示后疫情时代人们对商用洗手间的看法
Excel Dryer, Inc.与MetrixLab的研究表明
-
佳得乐与曼城球员共同揭晓 #THENOD 在足球场上的无声超凡力量
佳得乐 (Gatorade) 的广告牌现已正式出现在欧洲
-
默沙东与中国癌症基金会签署HPV疫苗捐赠协议
加速消除宫颈癌 共筑HPV健康防线 北京2024年11
-
维颜嘉西安国际医学中心新品发布会,中韩专家联袂推荐
杭州2024年11月24日 /美通社/ -- 2024年11月19日西安
-
"爱肺骑行,你我相伴"2024健康骑行公益活动圆满举行
上海2024年11月23日 /美通社/ -- 11月是国际肺癌关
-
-
-
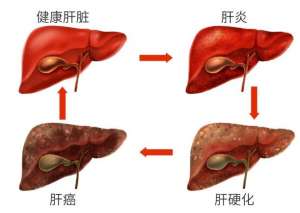
肝纤维化不能吃什么?肝纤维化饮食的注意事项有哪些?
中国人口14亿多,其中有9000万乙肝病毒感染者
-
肝硬化需要全疗程用药吗?哪个品牌的软肝片好?用药讲疗程,安全有保证!
人们常说“慢性病要以慢治慢”。古人倡导“
-
肝硬化的突出表现有哪些?用什么药软肝效果好?保肝软肝就选中成药
肝硬化是一种常见慢性病,引起肝硬化的病因
-
肝硬化的前期表现有哪些?肝硬化前期该如何选择软肝药品呢?
中国人的饮酒文化众所皆知,都特别喜欢酒桌
-
肝硬化患者吃什么药好,复方鳖甲软肝片有效吗?疗效值得认可
我国是肝病高发生率大国,每年有超过120万病
-
肝硬化吃复方鳖甲软肝片怎么样?复方鳖甲软肝片功效与作用来评价
肝硬化是一种常见的慢性肝病,是由一种或多
-
-
-
-
-
-
-
-
-
-
-
-
-